
Breast cancer is a malignant tumor that occurs in the glandular epithelial tissue of the breast, and is one of the most typical malignant diseases. Breast cancer is usually classified into five subtypes based on immunohistochemical markers, tumor grade, and proliferation level. This classification determines the means of treatment. However, due to cancer recurrence, drug resistance, or cancer metastasis, a large proportion of treatment methods are not successful. Therefore, the current classification system cannot fully capture the genetic and molecular status of cancer, and a refined classification system may be more conducive to predicting appropriate therapy and scope for different patients.
How Does SWATH-MS Improve Breast Cancer Classification
In July 2019, Ruedi Aebersold's research team published a research paper titled "Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry" in Cell Reports. In this study, SWATH-MS (DIA) technology was used to perform large-scale protein quantification of breast cancer tissues, and in-depth investigation of the five common subtypes of breast cancer was conducted. The study found that the consistency between protein subtypes and conventional classification subtypes reached 84%, revealing three key proteins that contributed most to the classification. Differences were also found between traditional tumor classification subtypes and protein level classification subtypes. These findings demonstrate that this technology can provide a refined classification of breast cancer at the protein level, and provide great help for the treatment strategy of breast cancer patients.

1. Establishment of a spectral library for quantitative analysis of breast cancer-associated proteins by SWATH-MS
The authors established a comprehensive breast cancer proteome spectral library by analyzing samples from breast cancer patients. The reference library contained 28,233 peptides and their modified variants ([FDR]<0.01), representing 4,443 proteins. This spectral library was used for quantitative analysis of proteins in breast cancer tissues identified by SWATH-MS (DIA) in subsequent experiments.
2. SWATH-MS analysis quantifies 2,842 proteins in 96 patient samples
The authors conducted proteomic analysis on tumor tissues from 96 breast cancer patients, which were classified into five conventional breast cancer subtypes based on their lymph node status, ER, PR, HER2 status, and tumor grade by pathologists. For each subtype, lymph node negative and lymph node positive samples were pooled separately, resulting in 10 sample pools. Using the SWATH (DIA) spectral library established above, the authors extracted quantitative data of 25,278 protein-specific peptides and their modified variants, representing 2,842 proteins in all individual samples. These 2,842 quantified proteins cover most of the known molecular processes related to breast cancer.
3. The comparison between protein subtypes and conventional breast cancer subtypes
The authors performed unsupervised clustering analysis on the 96 samples. In Figure 1A, lymph node-negative and -positive samples of each subtype were closely clustered together, indicating high reproducibility of the experiment. In Figure 1B, correlation clustering analysis was performed on the 96 samples, and the authors found that the clustering of protein subtypes was very close to that of traditional tumor subtypes. They also found that some subtypes had higher heterogeneity, indicating that traditional tumor classification may not have considered tumor heterogeneity.
Read more:phospho proteomic profiling
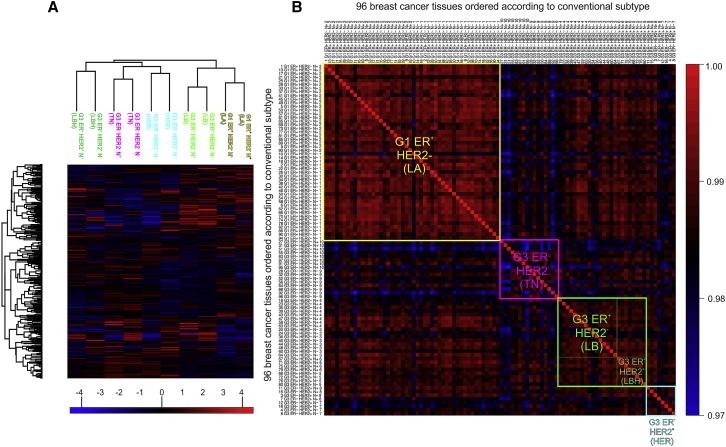
Figure1 | Study on the correlation between conventional subtypes and protein subtypes classification of breast cancer tissue
4. Screening and validation of key proteins
By analyzing the breast cancer-related protein pathways, key proteins for breast cancer subtypes were screened. After dividing the 96 samples into five conventional subtypes, a decision tree was established, which revealed a list of 22 key proteins (with some overlap in different comparisons). These 22 proteins in the decision tree contained three key nodes.
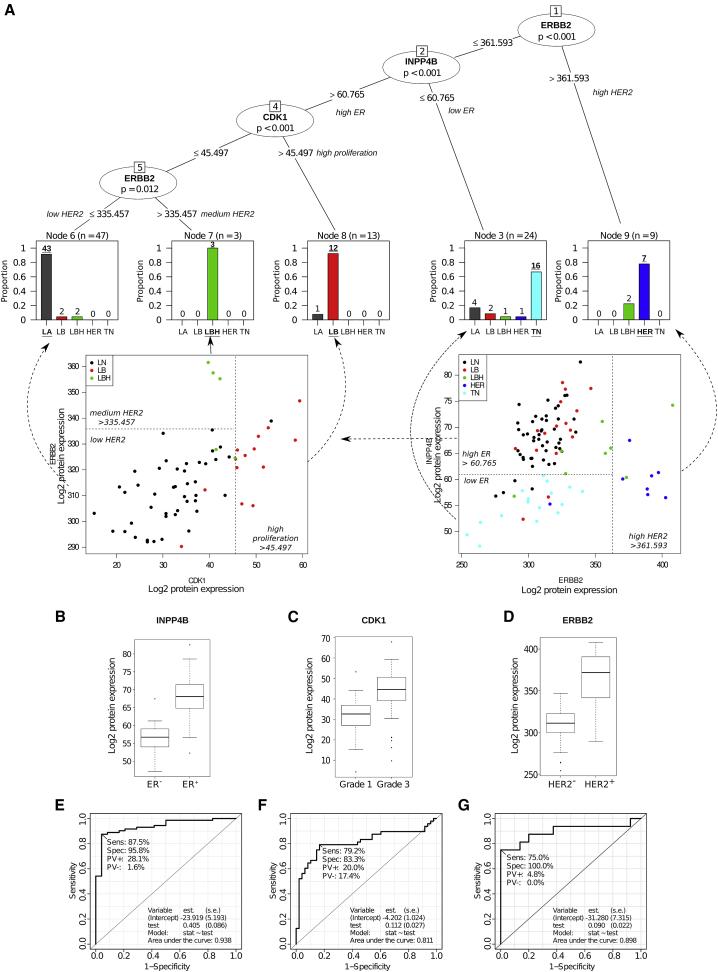
Figure2 | Classification of breast cancer patients based on tumor tissue protein levels.
The differential expression of these three proteins reflects the key clinical parameters of breast cancer subtypes: ER status, tumor grade, and HER2 status. Using the decision tree, breast cancer protein subtypes were classified with 84% concordance with traditional classification. Furthermore, statistical analysis of the key proteins in the 96 samples and in 116 samples reported in other studies showed significant differences in all three key node proteins, confirming that protein-level breast cancer subtype classification is more accurate and reliable than traditional classification.
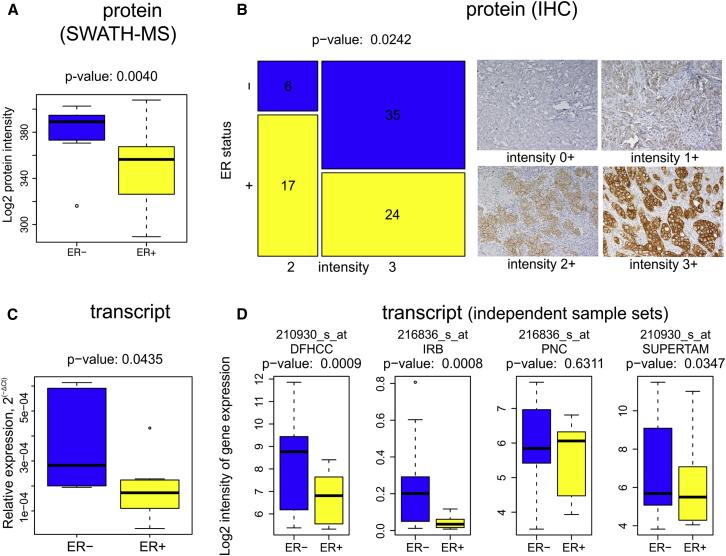
Figure 3 | The expression of ERBB2 protein and transcription in ER-/HER2+ and ER+/HER2+ breast cancer tissues
The author conducted a correlation analysis of fold changes at the protein and transcriptome level for 883 breast cancer patients worldwide. First, they performed a correlation analysis of fold changes for 2,782 proteins obtained from SWATH-MS (DIA), and the overall fold changes ranged between R=0.17 to R=0.29. Then, they analyzed the fold changes of the three key proteins identified in the decision tree, which ranged between R=0.67R=0.81. Although the global fold changes of total proteins were relatively low, the fold changes of the key proteins were high, indicating the importance of studying breast cancer at the protein level and classifying breast cancer subtypes based on protein expression.
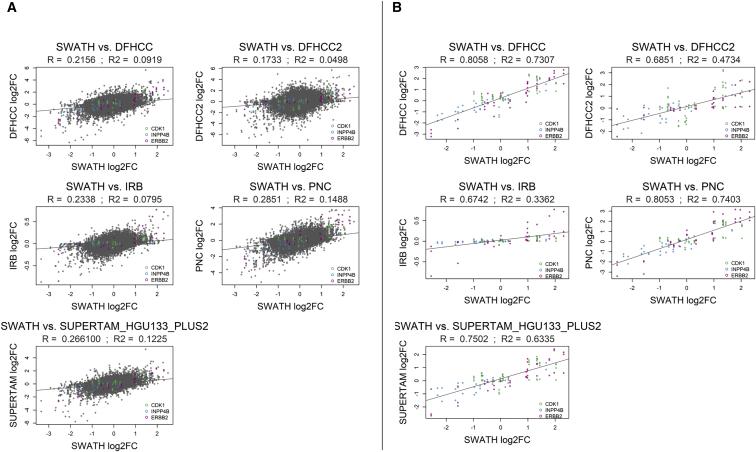
Figure 4 | Correlation analysis of the protein levels
Conclusion
This study explored and confirmed the applicability of SWATH-MS (DIA) for protein subtyping of human tumor samples under relatively high throughput conditions. The results showed that protein-based classification was more effective than traditional classification in resolving breast cancer subtypes and has the potential to improve traditional classification. In addition, it also demonstrated the great potential of data-independent acquisition methods (such as SWATH-MS) for research. This method of tissue classification is not limited to breast cancer and is effective for classifying other diseases and clinical specimens. Although we are not yet at the stage of making clinical decisions based on protein subtyping data, this study can further stimulate the research process and obtain more comprehensive treatments and better clinical outcomes. The SWATH spectrum library of breast cancer and high-quality proteomics data of 96 breast tumors will provide valuable resources for future protein marker research.
This study investigated the accuracy of SWATH-MS (DIA) for qualitative and quantitative analysis of breast cancer tissue under high-throughput conditions, demonstrating the powerful potential of this method and confirming the feasibility of protein-based classification for breast cancer subtypes, providing new directions for the diagnosis and treatment of breast cancer.
Reference:
- Ruedi Aebersold et al,. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. cell reports 2019








